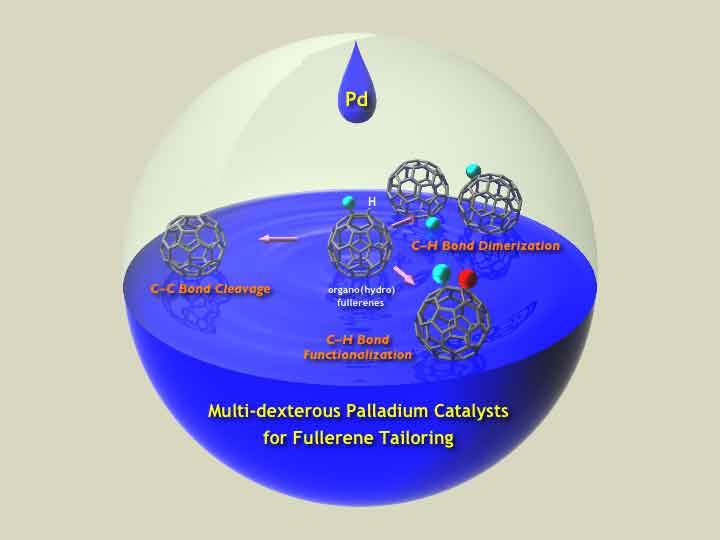
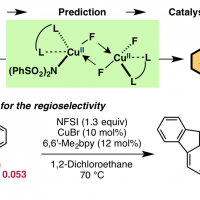
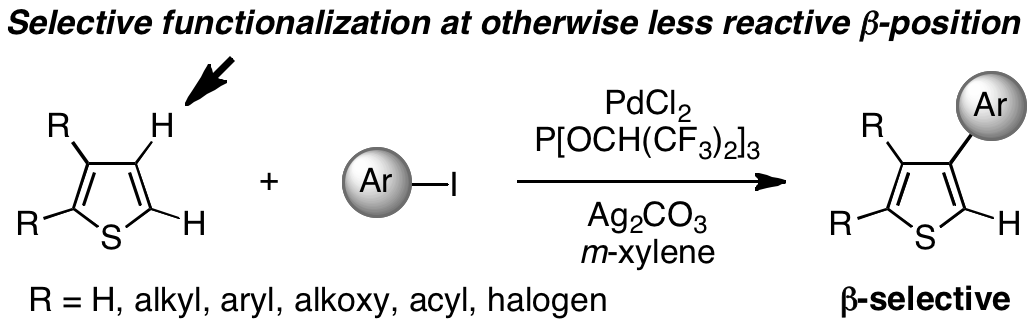
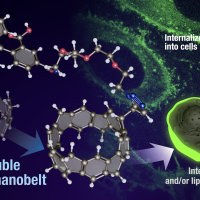
日本語
日本語
Y. Segawa, H. Omachi, K. Itami
Org. Lett. 2010, ASAP. DOI: 10.1021/ol1006168
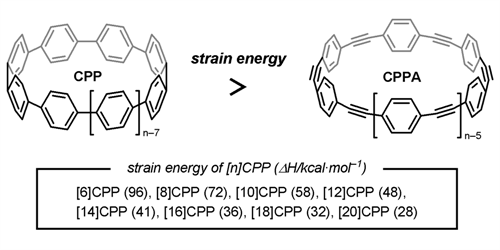
The structures and strain energies of cycloparaphenylenes (CPPs) have been determined by DFT calculation at the B3LYP/6-31G(d) level of theory. Fifteen stable conformations of [12]CPP were found as local minimum structures. It was also found that benzene rings of [12]CPP can rotate rather freely at room temperature. The strain energies of [n]CPP (n = 6−20) were estimated on the basis of the homodesmotic reaction using CPP, biphenyl, and p-terphenyl. It was also found that CPPs have higher strain energy in comparison to cycloparaphenyleneacetylenes (CPPAs).