日本語
日本語
Brandon E. Haines, Yutaro Saito, Yasutomo Segawa, Kenichiro Itami, and Djamaladdin G Musaev
ACS Catal. 2016, 6, 7536–7546. DOI: 10.1021/acscatal.6b02317
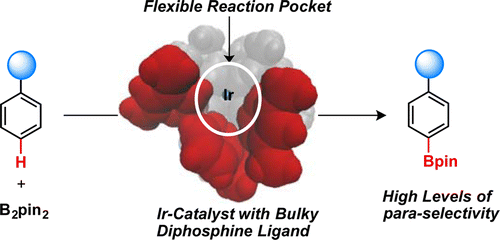
We used DFT calculations to elucidate the mechanism and source of regioselectivity for Ir-catalyzed para-selective C–H borylation with the bulky Xyl-MeO-BIPHEP diphosphine ligand (L1). We found that the bulky diphosphine–Ir complex forms a flexible reaction pocket that roughly mimics the role of an enzyme active site by modulating access of the substrate to the Ir center. It is shown that the regioselectivity of the reaction arises from a complicated balance of the attractive and repulsive interactions between the substrate and ligand and their corresponding entropic penalties across the high-energy C–H activation and C–B bond formation transition states in the reaction pocket. We predict a trend of increasing para-selectivity with an increase in the size of the substrate substituent in the order SiH3 (and Me) < SiMe3 < Si(t-Bu)3, which is consistent with the experimentally observed trend. It is shown that an increase in the steric bulk of the ligand by introducing bulkier 3,5-substituents to the phenyl rings of the diarylphosphino groups of L1 decreases the para-selectivity. This computational prediction was validated by experiments on C–H borylation of trimethyl(phenyl)silane by using the 3,5-di-tert-butylphenyl analogue of MeO-BIPHEP (L3) as a diphosphine ligand that showed a decrease in para/meta ratio from 88:12 (L1) to 50:50 (L3). Combination of the presented computational and experimental results illustrates that the regioselectivity of the reaction is not fully governed by repulsive steric interactions but instead by a complex balance of steric and electronic interactions between the substrate and flexible reaction pocket. We expect that the provided detailed mechanistic study will greatly enhance our ability to design the next generation of ligands with increased para-selectivity and generality.