
 日本語
日本語
Cristopher Camacho, Thomas A. Niehaus, Kenichiro Itami, and Stephan Irle
Chem. Sci., 2012, Advanced Article. DOI: 10.1039/C2SC20878D
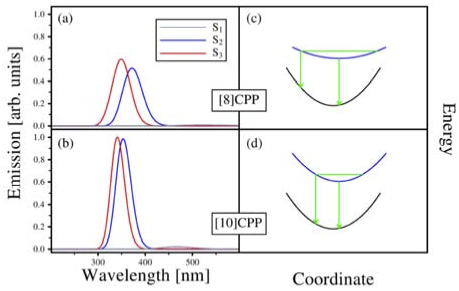
Quantum chemical electronic structure calculations were employed to investigate the nature of the low-lying excited states of [n]cycloparaphenylenes ([n]CPPs) and the role of geometrical distortions in the bright states. The lowest-energy bright states involve single-electron excitations from S0 ground state to S2 and S3 states, which are at the Franck-Condon geometry the two components of a twofold degenerate 1E state. They couple to a twofold degenerate e vibration which induce deformation of the CPP geometry from circular to oval shape. Non-radiative decay from the S2 states to the non-degenerate, dark S1 state is apparently suppressed due to symmetry rules and the extraordinarily high molecular symmetry of the CPP structure, and the emission spectral features in large n can therefore largely be attributed to the E x e Jahn-Teller system involving S2 and S3. However, absorption and emission energies computed at the respective S0 and S2/S3 minimum energy geometries are found to be nearly independent from the number of phenylene units n in the CPP molecules. In contrast, molecular dynamics simulations performed on the excited state potential surfaces are able to explain the experimentally observed fluorescence blueshift of the strongest emission peaks with increasing molecular size. This unusual feature turns out to be a consequence of generally higher structural flexibility in small [n]CPPs, causing greater Stokes shifts, while large [n]CPPs are generally more rigid and therefore feature smaller Stokes shifts (“dynamic blueshift”). For the same reasons, symmetry rules are violated to a greater extent in small [n]CPPs, and it is expected that a “static blueshift” due to emission from S1 becomes enhanced.



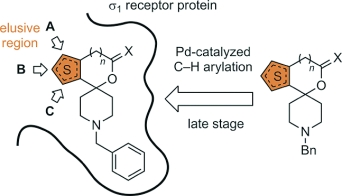
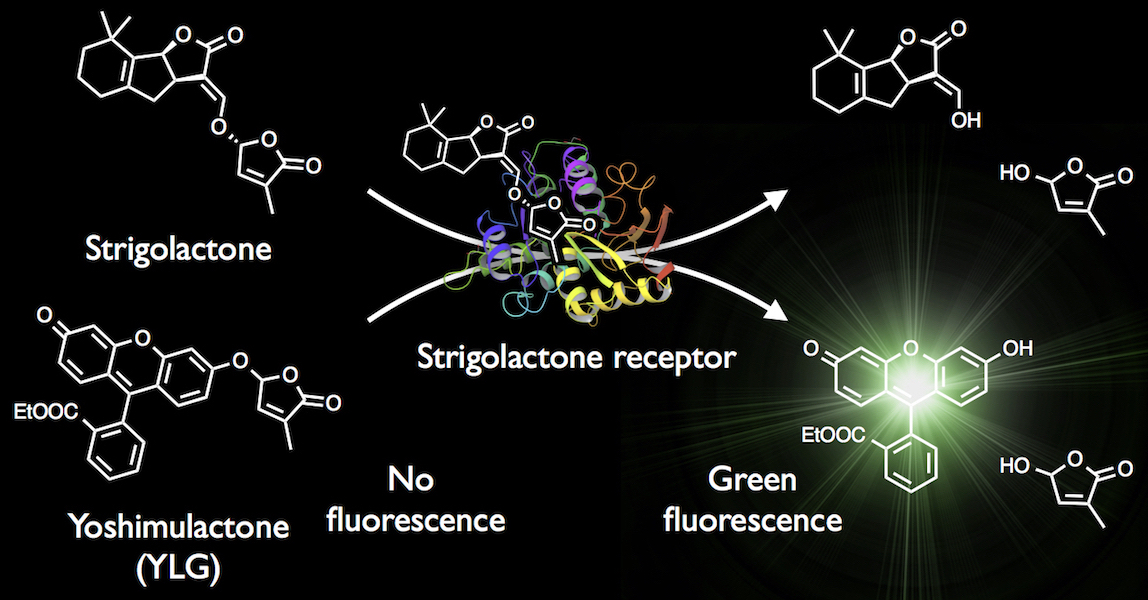




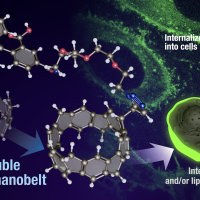















{kind=link}